RiboSketch
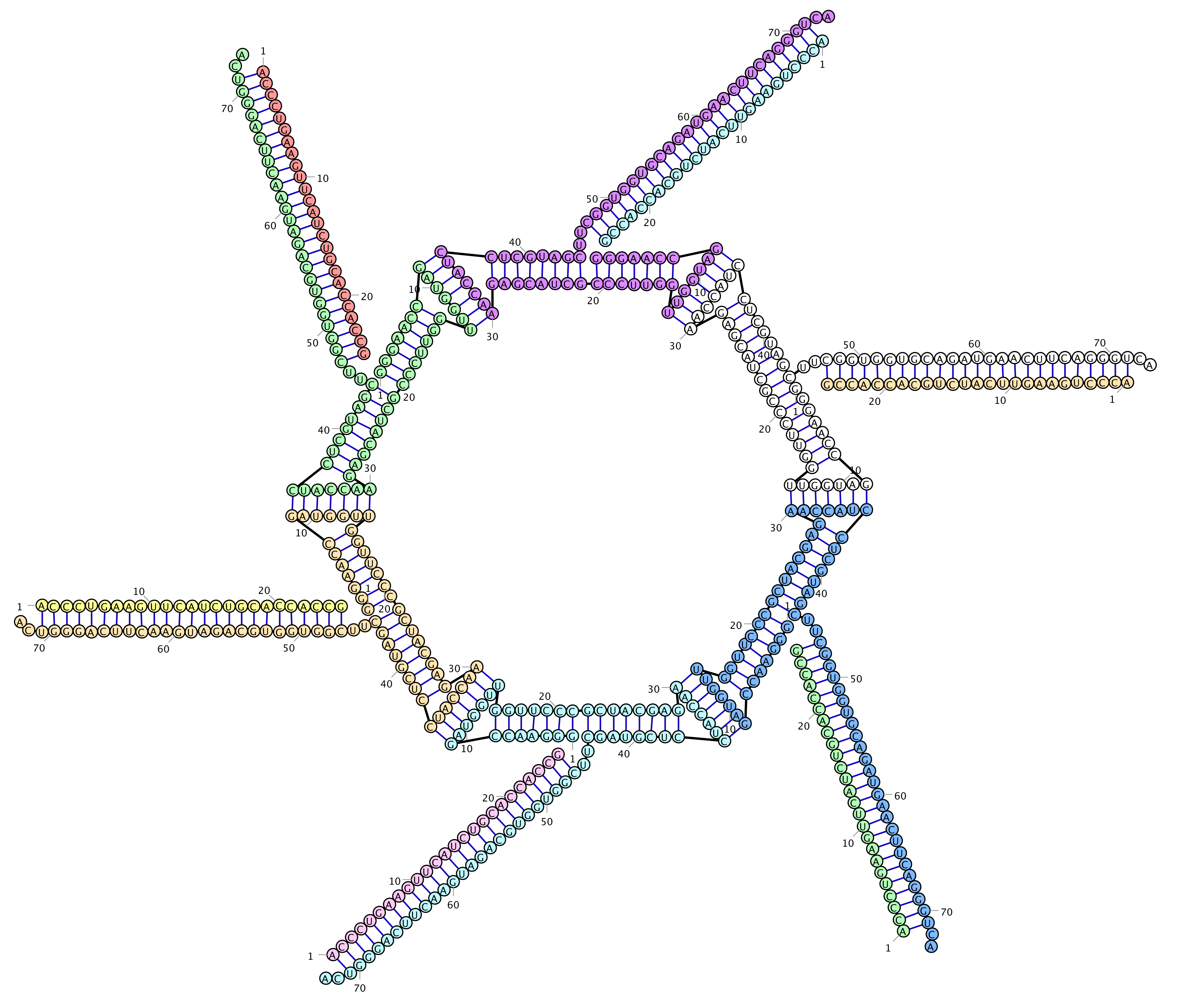
Bond editing, E. Coli 5S rRNA with SHAPE data coloring

Bioinformatics Publication
If you use RiboSketch in your scientific work, we would greatly appreciate a citation to our publication.
What is RiboSketch?
- RiboSketch is a drawing program for the production of RNA and DNA secondary structure images.
- The user provides an input file (.ct, .bpseq, .dbn, or the save file type .rs) containing the sequence and base-pairing of the strand(s).
Features
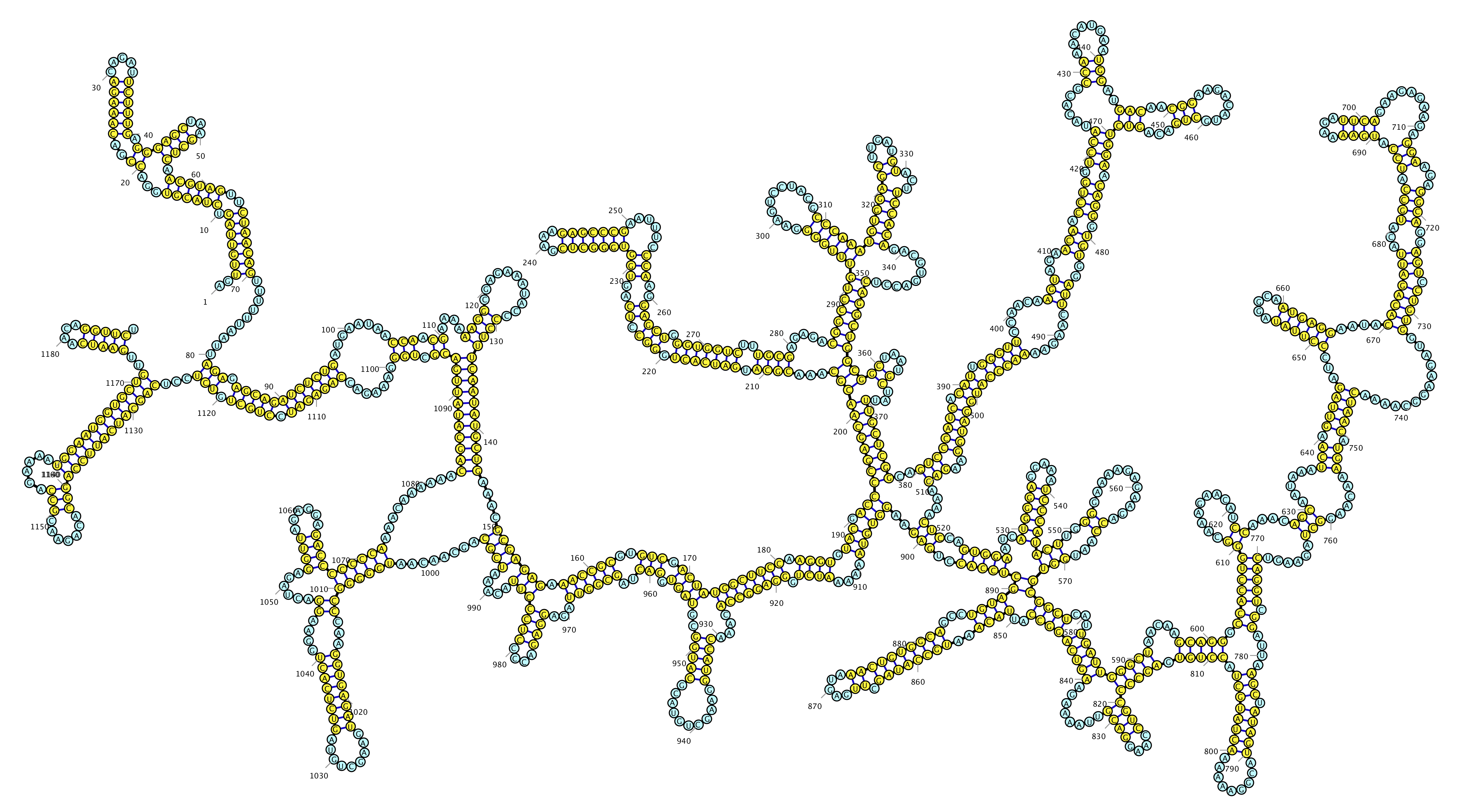
- Works with multiple strands, non-canonical interactions, hybrid RNA-DNA base-pairing, pseudoknots, and interstrand interactions
- Creates automatic layouts and circle diagrams
- Dynamic simulation mode
- Interactive editing is enabled through precise commands
- The user may save the state of the program and load previous save-states
- Actions may be undone and redone
Using the Program
- Load a secondary structure file (you can add additional bonds and non-canonicals once in the program)
- Default start state is the “Radial Layout”.
- Mouse over the top of the screen to access the MENU and to view COMMANDS.
Mouse Commands
- Click on (or drag a selection box over) bases to select them. Hold shift to multi-select or toggle the selection of bases.
- Drag selected nucleotides to move them.
- To select individual nucleotides, hold ALT/OPTION and click.
- To select one half of a helix, you can either:
- Select the helix, then shift click on the side you don't want to unselect it.
- Click and drag a selection box over one side of the helix.
- ROTATION: “m” rotates clockwise about the mouse cursor, “n” counterclockwise. Hold shift to rotate with a smaller angle.
- Select the helix, then shift click on the side you don't want to unselect it.
- Click and drag a selection box over one side of the helix.
Menu Features
Sliders
- Base Size: Size of nucleotides
- Bond Length: Distance between base-paired nucleotides
- Chain Length: Effective only during simulation mode, controls distance between adjacent nucleotides
- Color Scheme: “Pastel”, “White”, "Light", “Bright”, “Grey", and special ones:
- Base Type: Each nucleotide type gets its own color
- Rainbow: Each individual base gets its own color
- Structure: Paired bases: Yellow, Unpaired: Blue
- Custom: User must input number values corresponding to each base
- Base Type: Each nucleotide type gets its own color
- Rainbow: Each individual base gets its own color
- Structure: Paired bases: Yellow, Unpaired: Blue
- Custom: User must input number values corresponding to each base
Buttons
- Save: Writes state of program into a text file (.rs), which can be loaded into RiboSketch.
- Load File: Read a new secondary structure file into the program.
- Load Bonds: Add new bonds from a text file of comma-separated entries with format: “firstID secondID bondType”
- Example: 3 20 cWW, 4 19 tHS, 6 30 cSS
- Load Colors: Color nucleotides based on a space-separated list of numbers (probing data) from a text file.
- Radial Layout: Position nucleotides with the NAView algorithm, expanded to accommodate multiple strands and pseudoknots.
- Circle Layout: Nucleotides are positioned in a circle with base pairs drawn as chords.
- Simulation Mode: Apply forces.
- Sim. Selected Mode: Only apply forces to selected nucleotides.
- Rigid Helices: Enforce right angles for helices.
- Rigid Loops: Circularize loops and straighten stems.
- Rigid Hairpins: Circularize hairpin loops. Only has effect when Rigid Loops is OFF.
- Zoom Reset: Return zoom to default.
- Outlines: Toggle drawing circles around nucleotides.
- Labels: Display numbering for every tenth nucleotide.
- PNG Screenshot: Saves a screen image to the folder of the input file.
- SVG Screenshot: Saves an SVG file of the screen to the folder of the input file.
- Example: 3 20 cWW, 4 19 tHS, 6 30 cSS
Keyboard Commands
Editing
- ROTATION: “m” rotates clockwise about the mouse cursor; “n” counterclockwise. Hold shift to rotate with a smaller angle.
- "z": UNDO. Shift-Z to REDO.
- "s": Turn on or off SIMULATION (forces)
- "S": Toggle the SIMULATION MODE to apply forces to selected nucleotides only or all nucleotides.
- “f”: FLIP the halves of a selected helix.
- “x” or “y”: Flips the selected bases over the X or Y-axis.
- "r": RELAX FORCES for selected nucleotides. Effective only in Simulation mode.
Display
- + / - : Zooms the screen in or out
- 0 : Resets the view scale
- Arrows : When no nucletodies are selected, the arrow keys translate the window view.
- 1 / 2 : Brings the selected bases to the front or back, akin to Powerpoint
- b / B : Base SIZE +/- (Same as slider)
- o : Toggle OUTLINE
- i : Toggle LETTERS
- l : Toggle LABELS (Same as button)
- k : View base number in secondary file
- v : Toggle ANNOTATIONS (5' and 3', strand number)
- c : Changes the color scheme of the nucleotides
- p : Saves a PNG SCREENSHOT (Same as the button)
Adding and Deleting Bonds
- Adding: Hold Spacebar, click first base, click second base.
- Non-Canonical: Click "Load Bonds" button to add bonds of a specified type through text input (Ex: 3 25 cWW, 8 12 tHS)
- Deleting: Hold "d", click a base to delete its bonds.
The "RS" Data Format
- sequences: semicolon-separated list of RNA or DNA sequence strings which are in upper or lower case respectively
- starts: comma-separated list of zero-based indices of the first residue of each sequence
- positions: semicolon-separated string of x,y,z positions of each residue. In the current version the z-values are set to zero.
- radius: the radius of the residues in pixel units
- backbone_distance: target distance of two residues that are adjacent in sequence
- basepair_distance: target distance of two base paired residues
- color_scheme: an integer that stands for the color mode
- outline: true/false indicating whether the residues are drawn with a black circle
- movement: true/false indicating whether the simulation mode is on
- labels: true/false indicating whether the residue labels are shown or not
- rigid_helices: true/false indicating whether rigidifying helices is activated or not
- rigid_loops: true/false indicating whether rigidifying loops is activated or not
- rigid_hairpins: true/false indicating whether rigidifying hairpins is activated or not
- relax_ids: comma-separated list of residues whose basepairs are not affected by forces
- display_order: residues are displayed in this order. This is useful in order to achieve foreground/background effects
- pair_table: residue partners for each base pair (-1 for unpaired bases). Similar to base pair column in a CT format file.
- non_canonicals: extra information about non-canonical base pairs
- base_colors: assignable colors for each base (comma-separated RGB values between 0 and 1)